The genetics of our condition appear to mostly involve a genetic background involving microtubule-related genes (most significantly, those of the NBPF family) combined with a probable causative mutation in a gene related to the RAS/MAPK pathway.
In the NBPF family, increased copies of the CON1 subtype of the Olduvai domain in comparison to the HLS triplets has been correlated with reduced social capacity as well as increased brain size.
Given associations with autism spectrum disorder with megalencephaly and a significant and rather specific correlation in physiological features and symptoms in myself, as well as high paternal age, it is highly likely that a gain-of-function mutation in a RAS/MAPK pathway–related gene was causative for my severity of the condition.
Although my friend is also the product of high paternal age, he has much milder or absent RAS/MAPK pathway–related physiological features/symptoms (aside from autism spectrum disorder and megalencephaly). If he has a mutation in the RAS/MAPK pathway, it may be in a different part of the pathway to mine, leading to less pronounced physiological features.[1] For example, my features are especially reminiscent of Noonan syndrome, which is one of many RASopathies.
Microtubule-related genes
NBPF family – CON subtypes over HLS triplets
Although all six subtypes of the Olduvai domain contribute to brain size and the social mindset, it has been established that the HLS triplets have a greater contribution to the social mindset than the CON subtypes, while the CON subtypes have a greater contribution to brain size per copy than the HLS triplets.
Studies shown below found that in those with high-functioning autism spectrum disorder, higher copies of the CON1 subtype in comparison to the HLS triplets is associated with lower social capacity. These effects have been shown to be highly limited by sex, being significant only in males.
Autism spectrum disorder
A 2014 study examined repeat numbers of the CON1 subtype of the Olduvai domain in a sample of those with autism spectrum disorder (ASD). All had repeats in the normal range (56–88; mean: 70).[2]
The study found that a linear increase in number of repeats of CON1 was progressively correlated with more severe symptoms (those on the Autism Diagnostic Interview-Revised (ADIS-R): social reciprocity, communication and restricted/repetitive behaviours and interests). This was most prominent in social reciprocity.[2]
A 2015 follow-up study assessed repeat numbers of the CON1 subtype in another sample of those with ASD (mostly high-functioning). The repeats were in the normal range again (CON1: 54–78; mean: 66).[3]
The CON1 relationship was replicated with social reciprocity and communication but was no longer significant for restricted/repetitive behaviours and interests. Almost all of this sample were high-functioning and without intellectual disability, while the 2014 study’s sample contained more cases of autism with intellectual disability.[3]
The authors pointed out that the findings suggested more importance for Olduvai repeat numbers in those with higher-functioning and inherited autism than de novo cases involving intellectual disability, which are more often explained by rarer mutations.[3]
A 2019 study examined repeat numbers of CON1 and HLS1 in genomes of those with ASD, with HLS1 being used as a proxy for the HLS1–3 triplet. The study also factored in specific NBPF genes. The mean CON1 copy number of the sample was 70, and the mean HLS1 copy number was 200.[4]
In addition to the HLS1 association described on Genetics of the social mindset, in males, CON1 copy number was found to be incrementally correlated with impaired social reciprocity (0.18 points for each copy) and communication (0.13 points for each copy) scores on the ADIS-R. The associations were only found in inherited/familial autism and not de novo autism (with likely intellectual disability).[4]
In the sample, NBPF1 had 8.5–28.5 CON1 copies, and NBPF14 had 22.9–46.2 HLS1 copies.[4] The CON1 copy number of NBPF1 and the HLS copy number of NBPF14 were calculated to have explained 6.2% of the latent social phenotype.[4]
Possibly related to this are two things:
- NBPF1 has the most CON1 copies over HLS copies (with only around two HLS copies) of all the known NBPF genes as of 2012[5] and 2019.[6]
- Although NBPF14 does not have the most HLS Olduvai copies of any NBPF gene (though it is one of the four genes with the most HLS copies), it is adjacent to the most expressed of the three human-specific NBPF-paired NOTCH2NL genes, NOTCH2NLB.[7]
Brain size and intelligence
Brain size
A 2012 study showed that, in individuals with 1q21-deletion or -duplication-associated microcephaly or macrocephaly, Olduvai copy number showed the strongest association with brain size, especially with regards to the three conserved subtypes (CON1, CON2 and CON3). CON1 showed the strongest association, followed by CON3 and CON2. The finding was replicated in the form of an association with grey-matter volume in a healthy sample.[8]
Intelligence
A 2015 study examined repeat numbers of the CON1 and CON2 subtypes in association with IQ and mathematical ability in two population samples. The second sample had previously had a genetic analysis rule out an effect on IQ from many other genome-wide copy number variations. The CON2 copy number of most of the males ranged from 26 to 33, with a mean of 29.[9]
CON1 copy number was not found to have a significant association with IQ scores in this sample. However, in males, CON2 copy number was associated with higher IQ (as assessed on the Wechsler Intelligence Scale for Children) and higher mathematical ability (as assessed on the New Zealand Progressive Achievement Test for Mathematics). Each copy was associated with an average IQ-score increase of 3.3.[9]
The increases in IQ over other males of the same age rose proportionally with age before reaching a peak at a mean age of 11 years old and then plateauing. Head circumference at birth did not significantly correlate with future IQ.[9]
This was noted by the authors to correlate with the brain growth pattern found in those with high-functioning autism and/or higher intelligence.[9]
It may also correlate with reports of ‘progressive’ microcephaly, or microcephaly that becomes more apparent after birth, in loss-of-function mutations involving other microtubule-related proteins.[10][11][12]
There was no significant association found in females.[9]
Brain regions
In the aforementioned study, CON1 and CON2 copy number were found to increase the volumes and areas of all four bilateral lobes of the brain studied. Right-frontal-lobe surface area had the strongest association with both CON1 and CON2 copy number. The association was found to be slightly stronger with CON2 copy number. There were no CON1 or CON2 associations with white-matter volume or gyrification index (the amount of cortex buried within the sulcal folds vs. the amount visible on the outside of the brain).[9]
It was found that CON2’s effects on IQ remained substantial even after eliminating bilateral temporal surface area, right-frontal-lobe surface area and total grey-matter volume as factors. A portion of CON2’s association with IQ, however, was through its effects on bilateral temporal surface area. This contribution to IQ was larger than that of its effects on right-frontal-lobe surface area, despite the fact that it increased this area the most.[9]
Bilateral temporal surface area was found to correlate with a progressive increase in IQ, with left temporal surface area being slightly more important.[9]
The authors concluded that the Olduvai domain’s role in neural-stem-cell proliferation explained the effects of Olduvai dosage that could not be explained by brain region measurements, as neural-stem-cell proliferation also contributes to lobe surface area. However, they noted that the Olduvai domain also had effects on cortical thickness that appeared to be the result of mature neuron cell divisions.[9]
RAS/MAPK pathway–related genes
The RAS/MAPK pathway is a signalling pathway that contains small GTPases and protein kinases (enzymes) involved in signalling cell division and differentiation.
MAPK stands for mitogen-activated protein kinase, and mitogens or growth factors such as those of the fibroblast growth factor (FGF) family are signalling proteins for the RAS/MAPK pathway.[13]
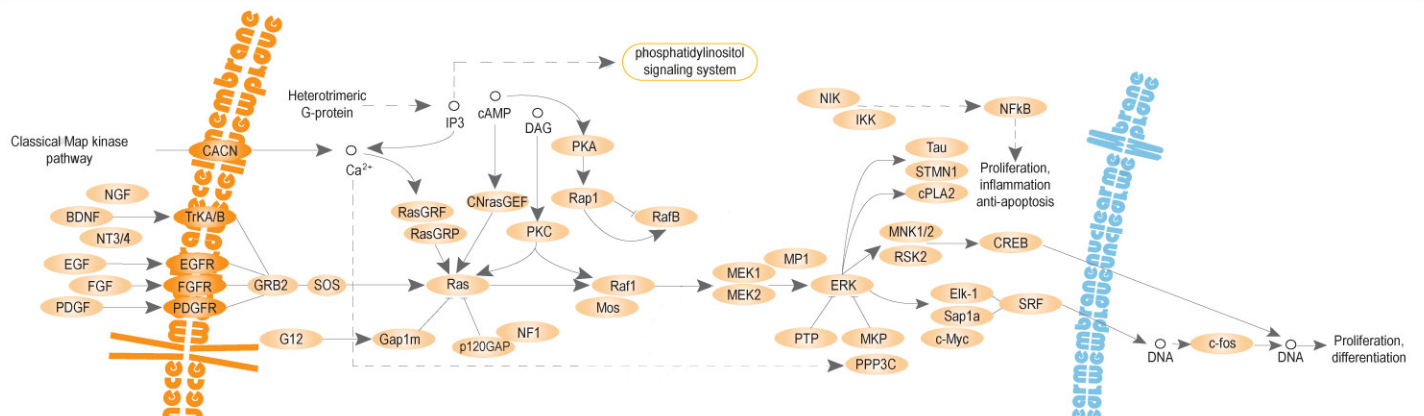
Source: [14]. Licence: CC-BY-4.0.
Paternal age effect
RAS/MAPK pathway–related genes constitute virtually all of the genes whose mutation inheritance is known to have a significant paternal age effect that surpasses simple accumulation of mutations in spermatogonia (sperm precursors) through repeated mitosis.[15][16]
Ordinarily, accumulation of mutations in spermatogonia over time has generally been found to result in a roughly twice increase in the risk of most genetic disorders in the children of fathers over age 40 compared to those around age 20.[17]
However, the effect in RAS/MAPK pathway–related genes is stronger, due to certain mutations in these genes conferring gain-of-function and increased dividing ability in the spermatogonia, which leads to a gradual increase in the population of spermatogonia with the mutation. This is also the basis of 95% of testicular tumours (germ cell testicular tumours).[15]
For rare conditions, this selectively advantageous population of spermatogonia has been reported not to increase the condition’s prevalence past 1 in 1,000[18] (e.g., 1 in 1,875 for achondroplasia with fathers aged 50 or older, compared to the typical 1 in 15,000,[19] an increase of 8 times), however it has been highlighted as a potential major factor in the commonality of neurodevelopmental disorders such as autism spectrum disorder.[15]
Conditions caused by mutations in Ras-related genes are collectively termed RASopathies.[15]
Some paternal-age-effect conditions and their respective gene mutations include the following:
- Protein tyrosine phosphatases:
- PTPN11 (gain-of-function)[20]
- Noonan syndrome (type 1 in OMIM)
- PTPN11 (gain-of-function)[20]
- Tyrosine kinases:
- RET (gain-of-function)[21]
- Multiple endocrine neoplasia types 2A and 2B
- RET (gain-of-function)[21]
- RAF kinases:
- Ras superfamily of small GTPases:
- Fibroblast growth factor receptors:
- Other:
Autism spectrum disorder with megalencephaly
RAS/MAPK pathway–related genes have been repeatedly linked to autism spectrum disorder, especially autism with megalencephaly, so much so that they have been called a ‘potentially unifying theory’ of autism.[13][24]
Autistic features are seen in Noonan syndrome,[25][26] neurofibromatosis type 1,[26][27] Costello syndrome[26] and cardiofaciocutaneous syndrome.[26][28] Macrocephaly is also seen in Noonan syndrome,[29] Costello syndrome[30] and cardiofaciocutaneous syndrome.[31]
Studies have shown the typical male bias in the autistic features among RASopathies,[26] including a 2017 study that reported in Noonan syndrome the same 5:1 male-to-female ratio seen outside the syndrome in a sample of 40 children with the condition.[28]
Paternal age effect in ASD
Like other genetic conditions, autism spectrum disorder is known to have a paternal age effect. However, if ASD had a paternal age effect linked to mutations affecting the RAS/MAPK pathway, one would expect it to have an increase in prevalence higher than 2 times among children of fathers aged 40+ compared to those of fathers aged around 20.
A 2006 study testing for a paternal age effect in a sample of those diagnosed with any autism spectrum disorder found an increase in risk of almost 6 times in fathers aged 40 or over compared to those aged 30 or under.[32]
A 2011 study focusing on infantile/childhood autism, excluding Asperger syndrome, found an increase in risk of 2.2 times in the children of fathers aged 50 or over compared to those aged under 30, similar to other non-RAS/MAPK mutations.[33]
A 2010 study included autism or Asperger syndrome (two thirds of the sample) and pervasive developmental disorder not otherwise specified (one third). It found risk increase of 2.58 for fathers aged 40 or over compared to those aged 25–29.[34]
Unlike high-functioning autism or Asperger syndrome, low-functioning autism without megalencephaly is often the result of any of a wide variety of rare loss-of-function mutations not necessarily related to the RAS/MAPK pathway, and the differing results are likely partly a result of the conflation of classic or low-functioning autism and high-functioning autism or Asperger syndrome into the results. This was noted by the authors of the 2011 study.[34]
However, it may also be that a genetic background (such as that involving the Olduvai-domain subtypes) found in families with a history of high-functioning autistic features or higher intelligence is necessary to render otherwise relatively silent RAS/MAPK-pathway mutations clinically significant in the children.
In these cases, the RAS/MAPK pathway–related mutation’s contribution to cell division may accentuate the relatively exclusive contribution to brain size and regional development (von-Economo-neuron areas vs. rest of the brain) from the Olduvai-domain subtypes.
In comparison to Noonan syndrome or other paternal-age-effect conditions, this may reduce the penetrance of the RAS/MAPK pathway–related mutation’s contribution to other RASopathy-like features (but not necessarily eliminate it, as represented by their presence in myself) to create our condition.
This is in addition to the fact that different mutations in different RAS/MAPK pathway–related genes will lead to differing phenotypes with more or less pronounced instances of certain RASopathy-like features.[1]
Other features
Aside from autism spectrum disorder and megalencephaly (and high paternal age), on Other features of our lack of the social mindset, I describe several other features of gain-of-function mutations in the RAS/MAPK pathway that I have.
They include mildly posteriorly rotated, mildly low-set ears, mild micrognathia, prominent volar pads, café au lait macules, gastrointestinal issues resembling dysmotility and delayed puberty.
These features are reflected in the known roles of the RAS/MAPK-pathway signalling protein fibroblast growth factor 8 (FGF8), also known as androgen-induced growth factor (AIGF).
Aside from brain development, FGF8’s signalling is known to be involved in mandibular/ear development (via the first branchial arch),[35] craniofacial neural crest cells,[35] pituitary development[36] and gonadal development.[35]
Volar pads
Volar pads are growths on the tips of the digits and the hands that appear at around 7–8 weeks of foetal development and are later caught up by growth in the rest of the digits and hand.[37]
Loss-of-function mutations in the serine/threonine kinase RPS6KA3 (which results in Coffin–Lowry syndrome[38]) and the GTPase RAB11B are associated with tapering fingers and microcephaly.[39][40]
One study found that the temporal-lobe region was particularly abnormal in Coffin–Lowry syndrome,[41] which may be linked to my friend’s lack of RAS/MAPK-pathway gain-of-function–related features and his synaptic differences in the temporal lobe.
Tapering fingers resulting from loss-of-function in the aforementioned RAS/MAPK pathway–related genes, which may be an inverse effect to the prominent volar pads associated with gain-of-function, are shown in the following photo for reference.
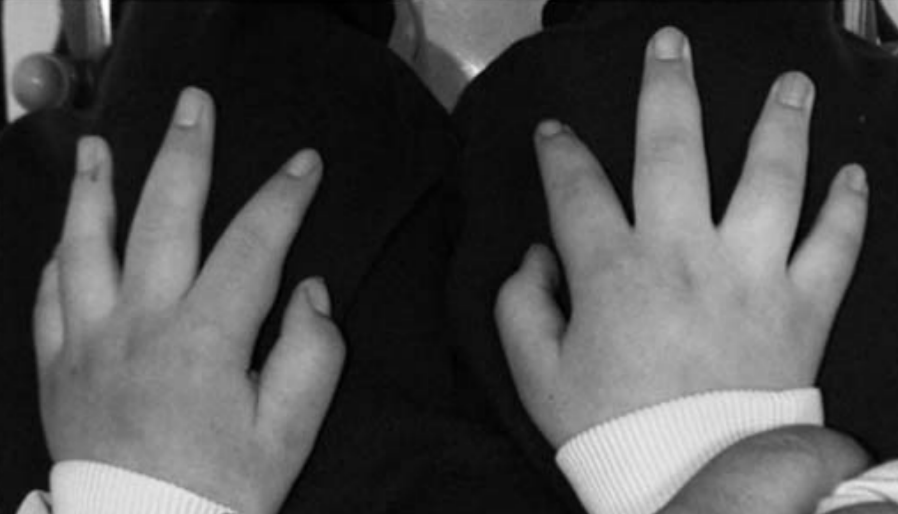
Source: [42]. Licence: CC-BY-4.0.
Pituitary gland
A 2019 study found statistically significant reduced pituitary volume in a small sample of those with Noonan syndrome type 1 and suggested that it contributed to the short stature in the syndrome.[43]
On the other hand, a 2016 study found that overexpressing BRAF (the cardiofaciocutaneous-syndrome gene) in the pituitary caused hyperplasia but disrupted differentiation, leading to hypopituitarism.[44] A 2017 study repeated this finding and found BRAF activating mutations in those with features of cardiofaciocutaneous syndrome and hypopituitarism.[45]
Neural crest
Genes related to the RAS/MAPK pathway, such as the RET and KIT receptor tyrosine kinases, are known to be involved in neural-crest-cell proliferation.
Neural crest cells derive from the neural tube and give rise to mid-facial cartilage, peripheral enteric nerves and melanocytes.
Loss-of-function mutations in RET are linked to Hirschprung’s disease (a congenital lack of nerves in the intestines),[46] while loss-of-function mutations in KIT are linked to piebaldism (patchy depigmentation),[46] an inverse effect to the café au lait macules associated with RASopathies.
References
- ^ a b Bentires-Alj, Mohamed; Kontaridis, Maria I.; Neel, Benjamin G (2006-03). "Stops along the RAS pathway in human genetic disease". Nature Medicine. 12 (3): 283–285. doi:10.1038/nm0306-283. ISSN 1546-170X.
- ^ a b Davis, Jonathan M.; Searles, Veronica B.; Anderson, Nathan; Keeney, Jonathon; Dumas, Laura; Sikela, James M (2014-03-20). "DUF1220 Dosage Is Linearly Associated with Increasing Severity of the Three Primary Symptoms of Autism". PLoS Genetics. 10 (3). doi:10.1371/journal.pgen.1004241. ISSN 1553-7390. PMC 3961203. PMID 24651471.
- ^ a b c Davis, JM; Quick, VB Searles; Sikela, JM (2015-6). "Replicated linear association between DUF1220 copy number and severity of social impairment in autism". Human genetics. 134 (6): 569–575. doi:10.1007/s00439-015-1537-6. ISSN 0340-6717. PMC 5886748. PMID 25758905.
- ^ a b c d Davis, Jonathan M.; Heft, Ilea; Scherer, Stephen W.; Sikela, James M (2019-02-15). "A Third Linear Association Between Olduvai (DUF1220) Copy Number and Severity of the Classic Symptoms of Inherited Autism". American Journal of Psychiatry. 176 (8): 643–650. doi:10.1176/appi.ajp.2018.18080993. ISSN 0002-953X.
- ^ O’Bleness, Majesta S.; Dickens, C. Michael; Dumas, Laura J.; Kehrer-Sawatzki, Hildegard; Wyckoff, Gerald J.; Sikela, James M (2012-09-01). "Evolutionary History and Genome Organization of DUF1220 Protein Domains". G3: Genes|Genomes|Genetics. 2 (9): 977–986. doi:10.1534/g3.112.003061. ISSN 2160-1836. PMC 3429928. PMID 22973535.
- ^ Fiddes, Ian T.; Pollen, Alex A.; Davis, Jonathan M.; Sikela, James M (2019). "Paired involvement of human-specific Olduvai domains and NOTCH2NL genes in human brain evolution". Human Genetics. 138 (7): 715–721. doi:10.1007/s00439-019-02018-4. ISSN 0340-6717. PMC 6611739. PMID 31087184.
- ^ Suzuki, Ikuo K.; Gacquer, David; Heurck, Roxane Van; Kumar, Devesh; Wojno, Marta; Bilheu, Angéline; Herpoel, Adèle; Lambert, Nelle; Cheron, Julian; Polleux, Franck; Detours, Vincent; Vanderhaeghen, Pierre (2018-05-31). "Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation". Cell. 173 (6): 1370–1384.e16. doi:10.1016/j.cell.2018.03.067. ISSN 0092-8674, 1097-4172. PMID 29856955.
- ^ Dumas, Laura J.; O’Bleness, Majesta S.; Davis, Jonathan M.; Dickens, C. Michael; Anderson, Nathan; Keeney, J. G.; Jackson, Jay; Sikela, Megan; Raznahan, Armin; Giedd, Jay; Rapoport, Judith; Nagamani, Sandesh S. C.; Erez, Ayelet; Brunetti-Pierri, Nicola; Sugalski, Rachel; Lupski, James R.; Fingerlin, Tasha; Cheung, Sau Wai; Sikela, James M (2012-09-07). "DUF1220-Domain Copy Number Implicated in Human Brain-Size Pathology and Evolution". The American Journal of Human Genetics. 91 (3): 444–454. doi:10.1016/j.ajhg.2012.07.016. ISSN 0002-9297, 1537-6605. PMID 22901949.
- ^ a b c d e f g h i Davis, Jonathon M.; Searles, Veronica B.; Anderson, Nathan; Keeney, Jonathon; Raznahan, Armin; Horwood, L. John; Fergusson, David M.; Kennedy, Martin A.; Giedd, Jay; Sikela, James M (2015-1). "DUF1220 copy number is linearly associated with increased cognitive function as measured by total IQ and mathematical aptitude scores". Human genetics. 134 (1): 67–75. doi:10.1007/s00439-014-1489-2. ISSN 0340-6717. PMC 5898241. PMID 25287832.
- ^ "OMIM Entry - # 604804 - MICROCEPHALY 3, PRIMARY, AUTOSOMAL RECESSIVE; MCPH3". Online Mendelian Inheritance in Man. (Archive version from 7 October 2020.)
- ^ "OMIM Entry - # 210720 - MICROCEPHALIC OSTEODYSPLASTIC PRIMORDIAL DWARFISM, TYPE II; MOPD2". Online Mendelian Inheritance in Man. (Archive version from 7 October 2020.)
- ^ "Strømme Syndrome | Hereditary Ocular Diseases". Hereditary Ocular Disease Database. (Archive version from 5 August 2020.)
- ^ a b Packer, Alan (2012-03-13). "SFARI | RAS pathway, a potentially unifying theory of autism". SFARI.
- ^ Kosigrim (2007-05-27). "The mitogen-activated protein kinase (MAPK) pathway, designed and donated to Wikpedia by en:Abgent, graphic execution by en:User:kosigrim". Wikimedia Commons. (Image cropped by myself.)
- ^ a b c d e f Goriely, Anne; Wilkie, Andrew O.M (2012-02-10). "Paternal Age Effect Mutations and Selfish Spermatogonial Selection: Causes and Consequences for Human Disease". American Journal of Human Genetics. 90 (2): 175–200. doi:10.1016/j.ajhg.2011.12.017. ISSN 0002-9297. PMC 3276674. PMID 22325359.
- ^ Forster, Peter; Hohoff, Carsten; Dunkelmann, Bettina; Schürenkamp, Marianne; Pfeiffer, Heidi; Neuhuber, Franz; Brinkmann, Bernd (2015-03-22). "Elevated germline mutation rate in teenage fathers". Proceedings of the Royal Society B: Biological Sciences. 282 (1803). doi:10.1098/rspb.2014.2898. ISSN 0962-8452. PMC 4345458. PMID 25694621.
- ^ Girard, Simon L.; Bourassa, Cynthia V.; Lemieux Perreault, Louis-Philippe; Legault, Marc-André; Barhdadi, Amina; Ambalavanan, Amirthagowri; Brendgen, Mara; Vitaro, Frank; Noreau, Anne; Dionne, Ginette; Tremblay, Richard E.; Dion, Patrick A.; Boivin, Michel; Dubé, Marie-Pierre; Rouleau, Guy A (2016-10-10). "Paternal Age Explains a Major Portion of De Novo Germline Mutation Rate Variability in Healthy Individuals". PLoS ONE. 11 (10). doi:10.1371/journal.pone.0164212. ISSN 1932-6203. PMC 5056704. PMID 27723766.
- ^ Goriely, Anne; Lord, Helen; Lim, Jasmine; Johnson, David; Lester, Tracy; Firth, Helen V.; Wilkie, Andrew O. M (2010). "Germline and somatic mosaicism for FGFR2 mutation in the mother of a child with Crouzon syndrome: Implications for genetic testing in “paternal age-effect” syndromes". American Journal of Medical Genetics Part A. 152A (8): 2067–2073. doi:10.1002/ajmg.a.33513. ISSN 1552-4833.
- ^ McDonald, Edward J.; De Jesus, Orlando (2020). "Achondroplasia". PubMed. PMID 32644689.
- ^ Roberts, Amy E.; Araki, Toshiyuki; Swanson, Kenneth D.; Montgomery, Kate T.; Schiripo, Taryn A.; Joshi, Victoria A.; Li, Li; Yassin, Yosuf; Tamburino, Alex M.; Neel, Benjamin G.; Kucherlapati, Raju S (2007-01). "Germline gain-of-function mutations in SOS1 cause Noonan syndrome". Nature Genetics. 39 (1): 70–74. doi:10.1038/ng1926. ISSN 1546-1718.
- ^ a b Leoni, C.; Flex, E (2017-12-07). "Costello Syndrome: The Challenge of Hypoglycemia and Failure to Thrive". EBioMedicine. 27: 5–6. doi:10.1016/j.ebiom.2017.12.006. ISSN 2352-3964. PMC 5828550. PMID 29248509.
- ^ "OMIM Entry - # 115150 - CARDIOFACIOCUTANEOUS SYNDROME 1; CFC1". Online Mendelian Inheritance in Man. (Archive version from 10 December 2020.)
- ^ "OMIM Entry - # 162200 - NEUROFIBROMATOSIS, TYPE I; NF1". Online Mendelian Inheritance in Man. (Archive version from 10 December 2020.)
- ^ Wen, Ya; Alshikho, Mohamad J.; Herbert, Martha R (2016-04-07). "Pathway Network Analyses for Autism Reveal Multisystem Involvement, Major Overlaps with Other Diseases and Convergence upon MAPK and Calcium Signaling". PLOS ONE. PLoS Journals. 11 (4): e0153329. doi:10.1371/journal.pone.0153329. ISSN 1932-6203.
- ^ Pierpont, Elizabeth I.; Hudock, Rebekah L.; Foy, Allison M.; Semrud-Clikeman, Margaret; Pierpont, Mary Ella; Berry, Susan A.; Shanley, Ryan; Rubin, Nathan; Sommer, Katherine; Moertel, Christopher L (2018-06-18). "Social skills in children with RASopathies: a comparison of Noonan syndrome and neurofibromatosis type 1". Journal of Neurodevelopmental Disorders. 10 (1): 21. doi:10.1186/s11689-018-9239-8. ISSN 1866-1955.
- ^ a b c d e Adviento, Brigid; Corbin, Iris L.; Widjaja, Felicia; Desachy, Guillaume; Enrique, Nicole; Rosser, Tena; Risi, Susan; Marco, Elysa J.; Hendren, Robert L.; Bearden, Carrie E.; Rauen, Katherine A.; Weiss, Lauren A (2014-01). "Autism traits in the RASopathies". Journal of Medical Genetics. 51 (1): 10–20. doi:10.1136/jmedgenet-2013-101951. ISSN 1468-6244. PMC 4230531. PMID 24101678.
- ^ Eijk, S.; Mous, S. E.; Dieleman, G. C.; Dierckx, B.; Rietman, A. B.; de Nijs, P. F. A.; ten Hoopen, L. W.; van Minkelen, R.; Elgersma, Y.; Catsman-Berrevoets, C. E.; Oostenbrink, R.; Legerstee, J. S (2018). "Autism Spectrum Disorder in an Unselected Cohort of Children with Neurofibromatosis Type 1 (NF1)". Journal of Autism and Developmental Disorders. 48 (7): 2278–2285. doi:10.1007/s10803-018-3478-0. ISSN 0162-3257. PMC 5995999. PMID 29423604.
- ^ a b Garg, Shruti; Brooks, Ami; Burns, Amy; Burkitt‐Wright, Emma; Kerr, Bronwyn; Huson, Susan; Emsley, Richard; Green, Jonathan (2017). "Autism spectrum disorder and other neurobehavioural comorbidities in rare disorders of the Ras/MAPK pathway". Developmental Medicine & Child Neurology. 59 (5): 544–549. doi:10.1111/dmcn.13394. ISSN 1469-8749.
- ^ "OMIM Entry - # 615355 - NOONAN SYNDROME 8; NS8". Online Mendelian Inheritance in Man. (Archive version from 10 December 2020.)
- ^ Gripp, Karen W.; Rauen, Katherine A.; Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E.; Bean, Lora JH; Stephens, Karen; Amemiya, Anne (1993). "Costello Syndrome". PMID 20301680.
- ^ Roberts, A; Allanson, J; Jadico, S K; Kavamura, M I; Noonan, J; Opitz, J M; Young, T; Neri, G (2006-11). "The cardiofaciocutaneous syndrome". Journal of Medical Genetics. 43 (11): 833–842. doi:10.1136/jmg.2006.042796. ISSN 0022-2593. PMC 2563180. PMID 16825433.
- ^ Reichenberg, Abraham; Gross, Raz; Weiser, Mark; Bresnahan, Michealine; Silverman, Jeremy; Harlap, Susan; Rabinowitz, Jonathan; Shulman, Cory; Malaspina, Dolores; Lubin, Gad; Knobler, Haim Y.; Davidson, Michael; Susser, Ezra (2006-09-01). "Advancing Paternal Age and Autism". Archives of General Psychiatry. 63 (9): 1026–1032. doi:10.1001/archpsyc.63.9.1026. ISSN 0003-990X.
- ^ Hultman, C. M.; Sandin, S.; Levine, S. Z.; Lichtenstein, P.; Reichenberg, A (2011-12). "Advancing paternal age and risk of autism: new evidence from a population-based study and a meta-analysis of epidemiological studies". Molecular Psychiatry. 16 (12): 1203–1212. doi:10.1038/mp.2010.121. ISSN 1476-5578.
- ^ a b Sasanfar, Roksana; Haddad, Stephen A.; Tolouei, Ala; Ghadami, Majid; Yu, Dongmei; Santangelo, Susan L (2010-02-22). "Paternal age increases the risk for autism in an Iranian population sample". Molecular Autism. 1 (1): 2. doi:10.1186/2040-2392-1-2. ISSN 2040-2392.
- ^ a b c "OMIM Entry - * 600483 - FIBROBLAST GROWTH FACTOR 8; FGF8". Online Mendelian Inheritance in Man. (Archive version from 7 October 2020.)
- ^ Kelberman, Daniel; Rizzoti, Karine; Lovell-Badge, Robin; Robinson, Iain C. A. F.; Dattani, Mehul T (2009-12). "Genetic Regulation of Pituitary Gland Development in Human and Mouse". Endocrine Reviews. 30 (7): 790–829. doi:10.1210/er.2009-0008. ISSN 0163-769X. PMC 2806371. PMID 19837867.
- ^ U.S. Department of Justice (2011). "The Fingerprint Sourcebook".
- ^ "OMIM Entry - # 303600 - COFFIN-LOWRY SYNDROME; CLS". Online Mendelian Inheritance in Man. (Archive version from 7 October 2020.)
- ^ "OMIM Entry - * 300075 - RIBOSOMAL PROTEIN S6 KINASE, 90-KD, 3; RPS6KA3". Online Mendelian Inheritance in Man. (Archive version from 1 November 2020.)
- ^ "OMIM Entry - # 617807 - NEURODEVELOPMENTAL DISORDER WITH ATAXIC GAIT, ABSENT SPEECH, AND DECREASED CORTICAL WHITE MATTER; NDAGSCW". Online Mendelian Inheritance in Man. (Archive version from 7 October 2020.)
- ^ Kesler, Shelli R.; Simensen, Richard J.; Voeller, Kytja; Abidi, Fatima; Stevenson, Roger E.; Schwartz, Charles E.; Reiss, Allan L (2007-04-01). "Altered neurodevelopment associated with mutations of RSK2: a morphometric MRI study of Coffin–Lowry syndrome". Neurogenetics. Springer Link. 8 (2): 143–147. doi:10.1007/s10048-007-0080-6. ISSN 1364-6753.
- ^ Morino, Tadao; Ogata, Tadanori; Horiuchi, Hideki; Yamaoka, Shintaro; Fukuda, Mitsumasa; Miura, Hiromasa (2016-08). "Eight years of follow-up after laminectomy of calcium pyrophosphate crystal deposition in the cervical yellow ligament of patient with Coffin–Lowry syndrome: A case report". Medicine. journals.lww.com. 95 (31): e4468. doi:10.1097/MD.0000000000004468. ISSN 0025-7974.
- ^ Potenza, Robert; Benn, Max; El-Naghy, Zeyad; McGuirk, Liam; Patale, Tara; Krasnow, Nicholas; Weiler, Winston; Haigney, James; Thompson, Carl; Tenner, Michael; Romano, Alicia; Noto, Richard (2019-04-30). "SAT-447 Diminished Pituitary Volume May be Linked to Short Stature in Patients with Noonan Syndrome". Journal of the Endocrine Society. 3 (Suppl 1). doi:10.1210/js.2019-SAT-447. ISSN 2472-1972. PMC 6552469.
- ^ Gregory, Louise; Kypranou, Nikolina; Besser, Rachel; Vignola, Lilliana; Temple, Karen; Davis, Justin; Gevers, Evelien; Dattani, Mehul; Gaston-Massuet, Carles (2016-08-19). "The MAPK Effector BRAF is Essential for the Integrity of Hypothalamic-Pituitary Development and Deregulation of this Pathway Causes Congenital Hypopituitarism".
- ^ Kyprianou, Nikolina; Gregory, Louise; Lillina, Vignola Maria; Besser, Rachel; Marinelli, Eugenia; Gualtieri, Angelica; Scagliotti, Valeria; Davis, Shannon; Gevers, Evelien; Dattani, Mehul; Gaston-Massuet, Carles (2017-10-20). "The MAPK effector B-Raf is essential for hypothalamic-pituitary axis development and activating mutations in BRAF cause congenital hypopituitarism". doi:10.1530/endoabs.50.OC5.6.
- ^ a b Dinsmore, Colin J; Soriano, Philippe (2018-12-01). "MAPK and PI3K Signaling: at the Crossroads of Neural Crest Development". Developmental biology. 444 (Suppl 1): S79–S97. doi:10.1016/j.ydbio.2018.02.003. ISSN 0012-1606. PMC 6092260. PMID 29453943.
- ^ a b Davis, Jonathan M.; Searles, Veronica B.; Anderson, Nathan; Keeney, Jonathon; Dumas, Laura; Sikela, James M (2014-03-20). "DUF1220 Dosage Is Linearly Associated with Increasing Severity of the Three Primary Symptoms of Autism". PLoS Genetics. 10 (3). doi:10.1371/journal.pgen.1004241. ISSN 1553-7390. PMC 3961203. PMID 24651471.
{kind=link}